- From the Schulich School of Medicine and Dentistry, University of Western Ontario, London, Ont.
Hypertriglyceridemia refers to a fasting plasma triglyceride measurement that is increased, typically above the 95th percentile for age and sex — although additional quantitative or qualitative lipoprotein abnormalities can also be present.1,2 Patients can fluctuate between hypertriglyceridemic states: given an appropriate metabolic stress, mild or moderate hypertriglyceridemia can deteriorate into severe hypertriglyceridemia. Elevated plasma triglyceride concentrations contribute to increased risk of cardiovascular disease, both directly and because such elevations “keep bad company” with associated risk factors such as obesity, metabolic syndrome, proinflammatory and prothrombotic biomarkers, and type 2 diabetes mellitus.2 The increased risk of acute pancreatitis is an additional consideration when a patient's triglyceride level is very high (typically > 10 mmol/L).
The two main sources of plasma triglycerides (also known as triacylglycerol) are exogenous (i.e., from dietary fat) and carried in chylomicrons, and endogenous (from the liver) and carried in very-low-density lipoprotein (VLDL) particles. In capillaries within fat and muscle tissue, these lipoproteins and chylomicrons are hydrolyzed by lipoprotein lipase into free fatty acids. After a meal, over 90% of the circulating triglycerides originate in the intestine and are secreted in chylomicrons, whereas during periods of fasting, endogenous triglycerides secreted by the liver as VLDL predominate. The increase in plasma of triglyceride-rich lipoproteins results from increased production from the liver and intestine (by means of upregulated synthetic and secretory pathways) or through decreased peripheral catabolism (mainly from reduced lipoprotein lipase activity).
Classification
Hypertriglyceridemia can be divided into primary and secondary types. In this postgenome era, a classification system for triglyceride disorders should be based upon molecular diagnoses, but a molecular basis for primary hypertriglyceridemia has been found in less than 5% of cases — and for secondary cases, no genetic susceptibility component that is reproducible.1Most patients with hypertriglyceridemia have at least one secondary factor; nevertheless, not everyone with equivalent exposure to secondary factors develops equally severe dyslipidemia, which suggests a role for endogenous primary monogenic or polygenic susceptibility.
The Adult Treatment Panel III3 of the National Cholesterol Education Program has suggested 4 triglyceride strata in the context of assessment of risk of cardiovascular disease: normal (< 1.7 mmol/L), borderline high (1.7–2.3 mmol/L), high (2.3–5.6 mmol/L) and very high (> 5.6 mmol/L). An alternative scheme, the World Health Organization–supported Fredrickson system of hyperlipoproteinemia phenotypes, was at one time widely taught, but has fallen into disuse. Here we review hypertriglyceridemia using clinical descriptive names, with corresponding Fredrickson numerical types included (in parentheses) for crossreference with older literature.
Primary hypertriglyceridemia
Chylomicrons normally are cleared rapidly from plasma by lipoprotein lipase with apolipoprotein (apo) C-II as a cofactor. Familial chylomicronemia (hyperlipoproteinemia type 1, in the Fredrickson system) and primary mixed hyperlipidemia (type 5) are each characterized by the pathologic presence of chylomicrons after a 12–14-hour period of fasting. Clinical features observed in both familial chylomicronemia and primary mixed hyperlipidemia include eruptive xanthomata (Fig. 1A), lipemia retinalis (Fig. 1C), hepatosplenomegaly, focal neurologic symptoms such as irritability, and recurrent epigastric pain with increased risk of pancreatitis. Samples of lipemic plasma develop a creamy supernatant when refrigerated overnight (Fig. 1B); when the plasma is tested, fasting triglyceride measurements are typically above 10 mmol/L in cases of either familial chylomicronemia or primary mixed hyperlipidemia.
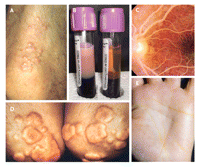
Fig. 1: Clinical manifestations of primary hypertriglyceridemia. A:Eruptive cutaneous xanthomas (here on a patient's knee) are filled with foam cells that appear as yellow morbiliform eruptions 2–5 mm in diameter, often with erythematous areolae. Most often associated with markedly elevated plasma chylomicrons in cases of familial chylomicronemia (hyperlipoproteinemia type 1) or primary mixed dyslipidemia (hyperlipoproteinemia type 5), they usually occur in clusters on the skin of the trunk, buttocks or extremities. B: Lipemic plasma. Whole blood has been allowed to stand at 4°C overnight. The sample on the left comes from a patient whose fasting total cholesterol result was 14.2 mmol/L and triglyceride concentration was 41.8 mmol/L. The sample on the right comes from a normolipidemic subject. C: Lipemia retinalis. A milky appearance of the retinal vessels and pink retina can be seen when plasma triglyceride concentration exceeds 35 mmol/L. D: Tuberous xanthomas, filled with foam cells, appear as reddish or orange, often shiny nodules, up to 3 cm in diameter. They are usually moveable and nontender. In patients with familial dysbetalipoproteinemia (hyperlipoproteinemia type 3), they usually appear on extensor surfaces; these are on a patient's elbows. E:Palmar crease xanthomas are filled with foam cells and appear as yellowish deposits within palmar creases. These skin lesions are pathognomonic for familial dysbetalipoproteinemia (hyperlipoproteinemia type 3). Photo by: Panels A, B and D are courtesy of Robert A. Hegele; panel C is courtesy of Ted M. Montgomery; and panel E is courtesy of Jean Davignon.
Key distinguishing features of familial chylomicronemia and primary mixed hyperlipidemia include initial manifestation during childhood for the former and in adulthood for the latter; biochemically proven deficiency of lipoprotein lipase, apo CII activity or homozygous gene mutations in the former, with less severe functional deficiency and infrequent detection of gene mutations in the latter; a much lower population prevalence of the former (about 1:106) than of the latter (about 1:103); frequent presence of secondary factors in the latter; and a greater elevation of total cholesterol in the latter, relative to that in familial chylomicronemia. In biochemical diagnosis, familial chylomicronemia features a loss of lipoprotein lipase activity in plasma collected after an intravenous dose of heparin; however, few laboratories still perform this test.
Familial hypertriglyceridemia (hyperlipoproteinemia type 4) is defined by an isolated elevation of VLDL, which is not as triglyceride-rich as chylomicrons are. This familial disorder has a population prevalence of some 5%–10%. Its molecular basis is still largely unknown but is likely to be polygenic, requiring a secondary factor for expression.1 Typically, patients with this disorder have moderately elevated plasma measurements of triglycerides (3–10 mmol/L), often with low levels of high-density lipoprotein–cholesterol (HDL-C). Familial hypertriglyceridemia is associated with increased risk of cardiovascular disease, obesity, insulin resistance, diabetes, hypertension and hyperuricemia.
The inheritance pattern of familial combined hyperlipoproteinemia (type 2B) is one of an autosomal dominant with variable penetrance, with a population prevalence of 2%–5%.4 The defining lipoprotein abnormalities are increased VLDL and low-density lipoprotein (LDL) with depressed HDL, associated with an abnormal lipoprotein profile in at least one first-degree relative. Affected people occasionally have obligate heterozygosity for LPL or APOC3 gene mutations, but the molecular basis underlying familial combined hyperlipoproteinemia is unknown in most instances.1 A recently defined gene that may be causative for this disorder is USF1, which encodes an upstream stimulatory factor,5 although several other genes (including APOA5 andAPOC3) have been variably claimed as causative.6
Finally, familial dysbetalipoproteinemia (hyperlipoproteinemia type 3) has a population prevalence of 1–2 in 20 000.7 The main observable lipoprotein abnormality is an increase in triglyceride-rich lipoprotein remnants, also known as intermediate-density lipoproteins or β-VLDL, which produce an equimolar elevation of plasma total cholesterol and triglyceride measurements.7 People with this disorder typically are homozygotic for the binding-defective APOE E2 isoform, which differs from the common E3 isoform by a substitution of cysteine for the normal arginine at residue 158 in the receptor-binding domain. Phenotypic expression, however, usually requires accompanying factors such as obesity, type 2 diabetes or hypothyroidism.7 Plasma levels of LDL are decreased because of interrupted processing of VLDL. An increased VLDL-C: triglyceride ratio and E2/E2 homozygosity are diagnostic. Affected people often have tuberous or tuberoeruptive xanthomata on the extensor surfaces of their extremities (Fig. 1D), planar-or palmar-crease xanthomata (Fig. 1E) and increased risk of cardiovascular disease.
Secondary hypertriglyceridemia
Some metabolic conditions are frequently (but not universally) associated with high triglyceride results, suggesting that people who develop secondary hypertriglyceridemia might have a subtle inherited metabolic defect that confers susceptibility. Obesity is probably the metabolic stressor most frequently associated with hypertriglyceridemia, although associations with poorly controlled type 2 diabetes and excessive alcohol consumption are also common.
Obesity, metabolic syndrome, diabetes. People with excess visceral adipose tissue often have elevated triglyceride and low HDL-C levels. About 80% of men with a waist girth of 90 cm or more and a plasma triglyceride level of 2 mmol/L or more typically have a metabolic triad of nontraditional cardiovascular-disease markers: hyperinsulinemia and increased levels of apo B and small, dense LDL particles. This triad can increase the risk of cardiovascular disease by up to 20 times.8
Impairment of the ability of insulin to stimulate glucose uptake and inadequate compensation for insulin insensitivity underlie type 2 diabetes. Moreover, among insulin-resistant people without type 2 diabetes, hyperinsulinemia is associated with a cluster of metabolic abnormalities called the metabolic syndrome.9 This syndrome, seen in people with central obesity, strongly predicts a future onset of type 2 diabetes. It is characterized by glucose intolerance, dyslipidemia (specifically, triglycerides > 1.7 mmol/L and low HDL-C concentrations) and hypertension.3 Hypertriglyceridemia, both in the metabolic syndrome and in type 2 diabetes, results from increased plasma concentrations of VLDL, with or without chylomicronemia;9 deficient lipoprotein lipase activity; increased cholesteryl ester transfer protein activity; and increased flux of free fatty acids to the liver.
A fatty liver is often associated with hypertriglyceridemia in people with obesity and insulin resistance. Several definitions of the metabolic syndrome exist,9 and it has been debated whether the clustered risk factors impart any risk above the simple sum of the individual components. However, the concept of the metabolic syndrome has proven to be useful in emphasizing the importance of obesity, insulin resistance and related lipoprotein disturbances in the assessment of cardiovascular disease risk. Hypertriglyceridemia of obesity, the metabolic syndrome and type 2 diabetes improves with weight loss and glycemic control.
Alcohol. Hypertriglyceridemia associated with alcohol intake also mainly results from increased plasma VLDL, with or without chylomicronemia. In some alcohol users, plasma triglyceride measurements can remain within the normal range because of an adaptive increase in lipolytic activity. However, alcohol can also impair lipolysis, especially when a patient has a pre-existing functional deficiency of lipoprotein lipase, which leads to markedly increased plasma triglycerides.
Renal disease. Although elevated LDL-C is the dominant abnormality, nephrotic syndrome is also characterized by increases in apo B–containing lipoproteins, including VLDL. The complex relation between renal disease and lipoprotein metabolism is reviewed in depth elsewhere,10 but the underlying mechanisms probably include overproduction by the liver, which concurrently increases albumin synthesis to compensate for renal protein wasting. Uremia is associated with elevated VLDL, which reflects impaired lipolysis, possibly from the toxic effect of uremic metabolites.
Pregnancy. During the third trimester of pregnancy, plasma triglyceride levels normally rise to as much as threefold,11 but this physiologic triglyceride increase has little clinical consequence. Marked triglyceride increases also result, however, when lipoprotein lipase activity is compromised. Although chylomicronemia during pregnancy is very rare, it can be complicated by pancreatitis, which can be fatal to both mother and fetus.12
Nonalcoholic fatty-liver disorder. This disorder may affect up to one-third of North Americans, which reflects the increasing prevalence of obesity, insulin resistance and metabolic syndrome.13,14 Among affected patients, up to one-third may also have nonalcoholic steatotic hepatitis.13,14 Lipotoxicity, oxidative stress, cytokines and proinflammatory mediators contribute to the progression from steatosis to nonalcoholic steatotic hepatitis. Elevated triglyceride and depressed HDL-C levels are the defining components of the dyslipidemia in nonalcoholic fatty-liver disorder. Small studies have indicated that treatment with statins is more effective than that with fibrates in correcting the dyslipidemia.15
Other medical conditions. Although hypothyroidism is usually associated with elevated LDL concentrations, triglycerides may also be elevated. Paraproteinemias (e.g., hypergammaglobulinemia in macroglobulinemia, myeloma, lymphoma and lymphocytic leukemias) and autoimmune disorders (e.g., systemic lupus erythematosis) can also cause hypertriglyceridemia, probably through immune-mediated interference of lipolysis.
Medications. Many drugs increase triglyceride concentrations (Box 1). If one is considered to cause hypertriglyceridemia, the indications for that medication should be reviewed. If dosage reductions, changes in route of administration or substitution with another class of medication are not practical, then marked elevations of triglycerides should be treated with diet or pharmacologic agents.

Patients taking highly active antiretroviral therapy, particularly protease inhibitors, frequently experience lipodystrophy, insulin resistance and dyslipidemia; up to 80% and 50% of patients develop hypertriglyceridemia and hypercholesterolemia, respectively.16 Combination highly active antiretroviral therapy was found to be associated with a 26% increase in relative risk of cardiovascular disease.17 Ritonavir and lopinavir are most strongly associated with dyslipidemias;16 3 reverse-transcriptase inhibitors, the nucleoside stavudine and the nonnucleoside nevirapine16 and efavirenz,18 less consistently so. Often, triglyceride levels can improve when agents are switched (if there is no compromise in antiretroviral efficacy).18,19 In one study,19 for instance, a change from a protease inhibitor to nevirapine or efavirenz reduced triglyceride levels by about 25%; the addition of pravastatin or bezafibrate further reduced them by about 40%.
Second-generation antipsychotic medications are known to be associated with obesity, hypertriglyceridemia,20 hyperglycemia and type 2 diabetes.21Clozapine and olanzapine disturb metabolism the most; risperidone and quetiapine have intermediate effects; and aripiprazole and ziprasidone, the fewest.20 Psychiatric disorders, because of associated lifestyles, may also predispose those affected to metabolic disturbances.22 Patients taking second-generation antipsychotics should be monitored regularly (every 8–12 months) for weight gain and changes in fasting plasma glucose and lipoprotein levels.21
Triglycerides and atherosclerosis
Moderate hypertriglyceridemia is almost certainly an independent risk factor for cardiovascular disease. The Prospective Cardiovascular Munster (PROCAM) study23 found increases in risk as triglyceride levels rose from 2.3 mmol/L to 9.0 mmol/L (after correction for other risk factors for cardiovascular disease). Other studies24 have shown a strong independent relation between plasma triglyceride levels and likelihood of cardiovascular disease. Meta-analyses of thousands of patients followed up for more than 10 years showed that a triglyceride elevation of 1 mmol/L increased risk of cardiovascular disease by 32% in men and 76% in women, independent of HDL-C levels.25
Complex mechanisms that underlie the association of triglycerides and atherosclerosis obscure the detection of any direct causal relation. Proatherogenic metabolic or biochemical abnormalities (e.g., obesity, type 2 diabetes, decreased levels of HDL-C, increased small dense LDL, increased free fatty acids, dysglycemia, hyperinsulinemia, increased plasma viscosity, increased inflammatory molecules, impaired fibrinolysis, prothrombosis) are often associated with elevated levels of triglycerides.2,24,25 Furthermore, triglyceride-rich lipoproteins and their remnants may directly contribute to the formation of arterial-wall foam cells. Chylomicrons are not directly atherogenic, although there are rare reports of atherosclerosis in patients with hyperchylomicronemia.26 In contrast, chylomicron remnants, VLDL and intermediate-density liproproteins are atherogenic.27 A newer concept is that postprandial lipemia is an independent predictor of cardiovascular disease,28although, since sample preparation and assay conditions are nonroutine, “postprandial” elevation of triglycerides has no operative definition.
Hypertriglyceridemia and pancreatitis
Hypertriglyceridemia increases the risk of acute pancreatitis, accounting for a minor but clinically relevant proportion of cases.29–32 Although a few patients can develop pancreatitis when their fasting triglyceride concentration is 5–10 mmol/L, its risk becomes clinically significant when fasting measurements exceed 10 mmol/L, a level at which chylomicrons are present.29–32 A threshold value of 1000 mg/dL (about 11.3 mmol/L) is often cited, but the clinical relevance and implications of a rounded threshold of 10 mmol/L for “very high” triglycerides are comparable. The capacity of the pancreas to produce an exocrine lipase might explain the specificity of pancreatic involvement.
Some patients with triglyceride-related pancreatitis probably have pre-existing abnormalities in their metabolism of lipoprotein.1 Pancreatitis risk is accentuated by any factor that can increase plasma triglycerides past 10 mmol/L. Triglyceride-induced pancreatitis can be preceded by episodic nausea and epigastric pain, during which serum amylase may not exceed common diagnostic cutoffs. Associated clinical clues include eruptive xanthomata or lipemia retinalis (Fig. 1).
The onset of full-blown acute pancreatitis can be prevented by restriction of dietary fat or the use of fibrates. Treatment of acute cases includes hemodynamic stabilization, cessation of all oral intake, placement of a nasogastric tube and control of metabolic disturbances. Restricted energy intake is associated with a brisk decrease in plasma triglyceride levels. Plasmapheresis has been attempted in extreme cases, but the benefit was transient. The threshold for developing pancreatitis can vary widely; some asymptomatic people live with triglyceride levels chronically in excess of 40 mmol/L.
Nonpharmacologic treatment
People with hypertriglyceridemia are frequently obese, insulin-resistant, hypertensive or diabetic, all of which are risk factors for cardiovascular disease. Treatment includes weight reduction, dietary modification and exercise. Dietary modification should decrease weight, overall energy intake, and intakes of fat and refined carbohydrates (i.e., foods with a high glycemic index).33 Alcohol consumption should be reduced or eliminated. In cases of severe hyperchylomicronemia, recommended fat intake is restricted to 10%–15% of total energy intake (about 15–20 g/d), with reductions in saturated, unsaturated and trans fats.3 Consultation with a dietician can be very helpful. For less severe cases of hypertriglyceridemia, restrictions to saturated and trans fat intake and increased aerobic activity can reduce plasma levels of triglycerides. The National Cholesterol Education Program advises a carbohydrate intake of 55%–60% and a protein intake of 15%–20% of daily dietary intake, whereas total and saturated fat should not surpass 30% and 7%, respectively.3 Plasma triglyceride response to diet and weight loss is about 25%,34 with marked variation among patients.34
Omega-3 fatty acids (e.g., eicosapentaenoic and docosahexaenoic acid) are components of both the Mediterranean diet and of fish oils.35 Daily consumption of 4 g of omega-3 fatty acids, along with with restricted energy and saturated-fat intakes, can reduce plasma triglyceride levels by as much as 20%. However, omega-3 fatty acids are rarely effective when used as the sole triglyceride-lowering therapy.
Pharmacologic therapy to lower the risk of cardiovascular disease
In general, monotherapy with a pharmacologic agent should be attempted first, together with dietary adjustments. Combination treatment may be required for refractory severe hypertriglyceridemia, but should be attempted only with caution and frequent monitoring of serum concentrations of creatine kinase, transaminase and creatinine.
Fibrates
Fibric acid derivatives such as gemfibrozil, bezafibrate and fenofibrate are a mainstay of hypertriglyceridemia treatment.36 These fibrates can reduce plasma triglyceride levels by up to 50% and raise plasma HDL-C concentrations as much as 20% (although these percentages vary). The complex mechanism of action of fibrates includes modulation of the activity of peroxisome proliferator–activated receptor-α in the liver, with reduced hepatic secretion of VLDL and increased lipolysis of plasma triglycerides.37Fibrates reduce the quantity of small, dense LDL particles and increase HDL-C.38 Fibrates can sometimes raise plasma LDL-C concentration, which in turn necessitates a change to another drug or the addition of a second agent. Fibrate therapy is generally very well tolerated, with very rare reports of it leading to hepatitis or myositis.36
The effectiveness of fibrates in reducing cardiovascular disease outcomes has been of long-standing concern. Earlier studies38–40 showed that fibrates reduced cardiovascular event rates; for instance, gemfibrozil resulted in a statistically significant benefit among men with high triglyceride and low HDL-C readings.40 The recent Fenofibrate Intervention and Event Lowering in Diabetes (FIELD) study found that plasma triglyceride, LDL and HDL-C levels in diabetic patients responded favourably to treatment with fenofibrate, but the reduction in the prespecified end point of cardiovascular disease (16%) was nonsignificant.41 Secondary and tertiary outcomes (e.g., nonfatal myocardial infarction, coronary revascularization, and progression of albuminuria and retinopathy) were significantly reduced. Serum creatinine was about 15% higher in the group taking fenofibrate and fell when fenofibrate was discontinued, which suggests a functional rather than a structural change.41However, subgroup analyses showed significant benefits of fenofibrate in patients without prior cardiovascular disease and after adjustment for “drop-in” statin use in the placebo group (i.e., increased use of prescribed statins by patients not treated with fenofibrate). Thus, enthusiasm for fibrate therapy is lower than that for statins, although selected patients might benefit.42
Statins
Statins are 3-hydroxy-3-methylglutaryl-coenzyme A reductase inhibitors. Newer statins used at higher doses can markedly reduce levels of triglycerides. They are not, however, a first-line therapy when triglyceride levels exceed 5 mmol/L.
An advantage of statins is the preponderance of evidence for reductions in coronary heart disease end points,43 especially among patients with type 2 diabetes.44 Like fibrates, statins are generally well tolerated and rarely cause myopathy or hepatic toxicity. Safety data from the FIELD study showed no rhabdomyolysis among the more than 1000 patients who took both fenofibrate and a statin.41 Potential beneficial effects of the combination of a statin and fibrate in the treatment of type 2 diabetes are currently being investigated in the ongoing Action to Control Cardiovascular Risk in Diabetes (ACCORD) study.45
Niacin
The daily consumption of up to 3 g of niacin (nicotinic acid) can lower plasma triglyceride levels by up to 45%, raise plasma HDL-C by up to 25%, and reduce plasma LDL-C by up to 20%.46 Older clinical trials suggested a reduction in cardiovascular events related to niacin treatment.47 However, niacin frequently causes light-headedness, cutaneous flushing, or pruritus. These adverse effects can be minimized by starting therapy at low doses then gradually increasing the daily dose; concomitant use of acetylsalicylic acid; or use of longer-acting preparations, such as Niaspan.48 Less common adverse effects include elevations of liver enzymes, increased levels of uric acid, gastrointestinal distress and worsened glucose tolerance.
Other lipid-lowering medications
Bile acid–binding resins worsen plasma triglyceride concentrations,36 whereas cholesterol-absorption inhibitors (e.g., ezetimibe) do not.49 A combination of fenofibrate and ezetimibe was recently shown to be safe and effective for patients with elevated triglyceride and LDL-C levels.50 Canada recently joined the United States in approving this combination for therapeutic use.
Emerging treatments
Rimonabant is a cannabinoid-1 receptor antagonist that decreases hunger and reduces food intake.51,52 Four phase-3 randomized, double-blind, placebo-controlled trials (the Rimonabant in Obesity trials, namely RIO–Europe, –Lipids, –North America and –Diabetes)52–56 have each evaluated rimonabant in various patient groups, showing successful, persistent reductions in obesity variables in the treatment groups, with concomitant improvements in metabolic biomarkers, including triglycerides.52–56 However, adverse effects such as depression and anxiety were also more common in those groups.54Nonetheless, rimonabant seems to be a promising new treatment for obesity and dyslipidemia.
Glitazar drugs are dual agonists of peroxisome proliferator-activated receptor-α (similar to fibrates) and -γ (similar to thiazolidinediones) and hold theoretical advantages for treatment of type 2 diabetes and metabolic syndrome. However, an analysis of phase 2 and 3 trials found significant associations between muraglitazar and death, myocardial infarction and stroke.56 This agent and others of the class have since been withdrawn from clinical use.
LPL gene therapy. Hyperlipoproteinemia type 1 caused by lipoprotein lipase deficiency causes recurrent and potentially fatal pancreatitis.1 The incidence of lipoprotein lipase deficiency can be up to about 1:5000 in regions of Eastern Quebec because of a genetic founder effect;57 in such populations, gene therapy is an interesting, if still theoretical, consideration. Management of lipoprotein lipase deficiency with fibrates is modestly effective. As for nonpharmaceutical treatments, most patients find it difficult to adhere to the recommended diet of less than 10% (of total energy intake from) fat. Even with aggressive management, plasma triglyceride levels in patients with familial chylomicronemia often remained over the 10 mmol/L threshold. Gene therapy, however, may help treat monogenic lipoprotein lipase deficiency,58 since lipoprotein lipase can be targeted to skeletal muscle: a modest increase in activity would then dramatically reduce plasma triglyceride levels. Using a “beneficial” allele of LPL, gene therapy has corrected elevated triglyceride levels in animals,59 which paves the way for trials involving people.
Target triglyceride goals
In 2006, guidelines from the Canadian Working Group on Hypercholesterolemia and Other Dyslipidemias no longer recommended a specific triglyceride target,60 whereas those from the National Cholesterol Education Program in the United States recommended that if triglyceride concentration exceeds 1.7 mmol/L, therapy should be aimed at attaining the LDL-C target specified for the patient's risk stratum of coronary heart disease.3 Table 1 presents a proposed strategy for management of patients whose triglyceride levels are elevated.

Conclusions
Moderate hypertriglyceridemia is a risk factor for cardiovascular disease; severe hypertriglyceridemia, for pancreatitis. Although reductions in plasma triglycerides probably reduce the risk of cardiovascular disease, the clinical trial evidence for fibrate monotherapy is weaker than that provided for reductions of plasma LDL-C concentrations. Nonetheless, improvement of triglyceride levels by control of secondary factors will help reduce risk of cardiovascular disease. The use of fibrates as prophylaxis against pancreatitis when plasma triglyceride results exceed 10 mmol/L is less contentious. In the next few years, new clinical trials (especially of statin–fibrate combination treatment) and the availability of new types of treatment may result in an evolution of the current approach to treating elevated plasma triglycerides.
Footnotes
- This article has been peer reviewed.Contributors: Khalid Al-Shali and Robert Hegele drafted the manuscript. George Yuan and Robert Hegele participated in the critical revision of the article. All authors have approved the final version for publication.Acknowledgements: We were supported by the Jacob J. Wolfe Chair in Functional Genomics, the Edith Schulich Vinet Canada Research Chair (Tier I) in Human Genetics, a Career Investigator award from the Heart and Stroke Foundation of Ontario and operating grants from the Canadian Institutes for Health Research, the Heart and Stroke Foundation of Ontario, the Ontario Research Fund, and Genome Canada through the Ontario Genomics Institute. George Yuan was supported by the University of Western Ontario Summer Research Training Program and by the Heart and Stroke Foundation of Ontario's Irwin Bernick Scholarship.Competing interests: None declared.
No comments:
Post a Comment